



- HYGEIA
- Services



All brain conditions are treated in close co-operation with the Departments of Oncology, Neurology, Internal Medicine, CAT scan and MRI, Radiosurgery and Pathology.
Below is a brief reference to the most common brain tumours, treated in the Neurosurgery Department (gliomas, meningiomas and pituitary adenomas).
Gliomas are primary, intracranial tumours usually stemming from one of the four types of glial brain cells: astrocytes (astrocytoma), oligodendrocytes (oligodendroglioma), ependymal cells (ependymoma) and finally neuroglia stem cells (glioma). Depending on their histology features, they are classified in 4 categories (grades) as per the World Health Organisation.
- Grade I: Pilocytic astrocytoma
- Grade II: Astrocytoma, ependymoma, oligodendroglioma
- Grade III: Anaplastic astrocytoma, anaplastic oligodendroglioma
- Grade IV: Glioblastoma multiforme
Grades reflect the aggressiveness of such tumours. While pilocytic astrocytoma does not have the predisposition to progress aggressively and its complete resection leads to cure, the same does not hold true for grades II-IV. Grade II tumours are characterised as low malignancy tumours. Through time however, they evolve most of the times to the most malignant forms of grade III and IV.
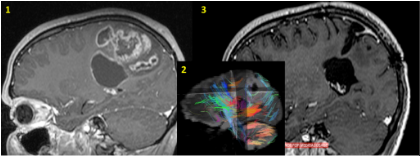
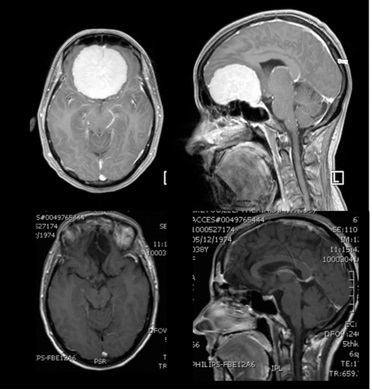
Glioblastoma (Grade IV as per WHO) of the right parietal lobe.
The patient presented with muscle weakness of the left upper extremity. The diffusion (insert image) MRI (image 1) has revealed a sizable lesion, its association with the brain’s motor centres and the pyramidal tract (insert image blue/light blue tracts), which transfers motoric orders to the body. The pre-operative imaging and the intra-operative neurophysiology imaging of such areas allowed for the macroscopically total resection of the tumour. During the post-operative course, complete remission of the symptoms was observed. The MRI (image 3) 2 years post-op, did not reveal any signs of relapse.
Mean survival depends on age, size and location of the tumour and ranges from 5-15 years. Grade III anaplastic astrocytoma is clearly more aggressive and develops more quickly, presenting a mean survival time from 0.5 to 5 years. Glioblastoma is the most common form of glioma, presenting a very rapid progression and the lowest mean survival time (0.5 – 2 years). The ependymoma is classified as grade II, it mainly develops in the brain’s ventricular system or along the spinal canal and at times leads to metastases through the cerebrospinal fluid. Anaplastic ependymomas are classified as grade III and require post-operative radiotherapy and chemotherapy. Oligodendrogliomas are classified as grade II, mainly develop in the frontal lobes forming cysts and calcifications. Following radical resection with/without radiotherapy, high mean survival time is observed (10-15 years). Under certain circumstances (1p19q gene mutations), it manifests excellent response to specialised chemotherapy protocols. The most malignant form is the anaplastic oligodendroglioma (grade III).
SIGNIFICANT CHANGES IN GLIOMAS CATEGORISATION / CLASSIFICATION
In the past decade, molecular profile genome studies have identified characteristic genetic changes associated with different glioma types. Such molecular features can nowadays be used to improve gliomas classification, to lead to individualised treatment and to improve patient prognosis.
Therefore, the World Health Organisation (WHO) tumours classification for the Central Nervous System (CNS) was revised in 2016 to incorporate molecular biomarkers together with the typical histology features into one integrated diagnosis, in order to determine glioma types as accurately as possible. This tactic led to the review of the histology examination, creating new glioma categories with common genetic features.
ROUTINELY USED DIAGNOSTIC BIOMARKERS
IDH1/IDH2 gene mutation codifying the human cytosolic NADPH- dependent isocitrate dehydrogenase, an enzyme involved in the citric acid cycle as a DNA repair enzyme, protecting normal cells from cancer cells, removing alkyl groups from the DNA is
- A diagnostic marker for diffuse grade II or III astrocytomas as well as for secondary glioblastomas (that have evolved from lower grade gliomas) and
- A good prognostic indicator (better response to chemotherapy).
1p/19q loss (co-deletion of chromosome strains 1p and 19q) is identified in more than 80% of grade II oligodendrogliomas as per WHO and in approximately 60% of grade III anaplastic oligodendrogliomas as per WHO. It is associated with better prognosis and better response to specific chemotherapy (PCV regime)
MGMT repair enzyme hypermethylation (O6-methylguanine-DNA-methyltransferase) that inhibits the function of this enzyme, renders glioblastoma or anaplastic oligodendroglioma cells particularly sensitive to treatment with temozolomide, an alkylating agent.
Other diagnostically relevant biomarkers include the loss of expression of nuclear ATRX, TERT promoter mutation, gene fusion KIAA1549-BRAF and BRAF-V600E and H3F3-G34 mutations.
NEW METHOD FOR THE CLASSIFICATION OF BRAIN GLIOMAS
The new CNS tumours classification by WHO in 2016 follows a multi-level approach for the classification of glioma, combining three layers of information to determine the definite, integrated diagnosis (highest level), as follows:
Highest level
Τελική (ολοκληρωμένη) ιστολογική διάγνωση που περιλαμβάνει όλες τις πληροφορίες
Level 3
Molecular information (diagnostic biomarkers testing results)
Level 2
Tumour malignancy histologic grade (WHO grade)
Level 1
Tumour histologic type
EXAMPLE Tumours of oligodendroglioma histologic subtype (level 1) and malignancy grade II WHO (level 2) in which IDH1/IDH2 gene mutation has been identified together with 10/q19 chromosome loss (layer 3) are given the definite (highest level) integrated histology diagnosis of ‘oligodendroglioma grade II WHO with mutated IDH1/IDH2 and 1p/19q chromosomal loss’. Such information facilitates the selection of the best post-operative treatment, as well as of potential treatment in case of future relapse.
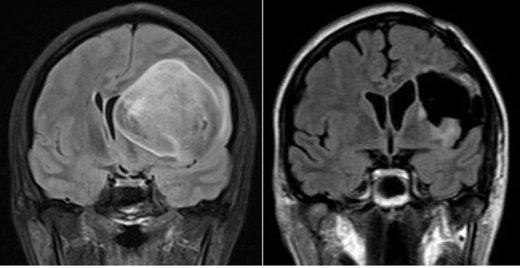
Cerebellar myeloblastoma
27 years old patient with symptoms of increased intracranial pressure and hydrocephalus due to obstruction of the brain’s 4th ventricle. Total excision via sub-occipital craniotomy. Image 2 presents the post-operative picture. The patient underwent Central Nervous System radiotherapy and chemotherapy.
Gliomas are diagnosed with the use of Brain Magnetic Resonance Imaging (MRI). In some cases, computed axial tomography is also necessary to determine tumour features (size, location). Additional imaging tests are in many cases required and serve specific purposes: Magnetic Spectroscopy, Functional MRI, Diffusion and perfusion techniques MRI and PET-CT scan.
The treatment of gliomas consists in the resection of the tumour through a craniotomy. Multicentric studies have confirmed that radical surgical tumour resection, resecting >90%, and the administration of radiotherapy and chemotherapy postoperatively, significantly improve life expectancy and quality of life for patients. Each case should be treated on an individual basis and in accordance with international protocols for glioma treatment. In certain cases of particularly unfavourable prognosis such as invasion of more than one brain lobes, extension in a functional centres area (speech, basal ganglia, stem, motor area), craniotomy does not offer any substantial benefit, since it does not offer radical tumour excision. In this case, confirmation of the diagnosis is usually recommended by means of histology examination, with stereotactic biopsy.
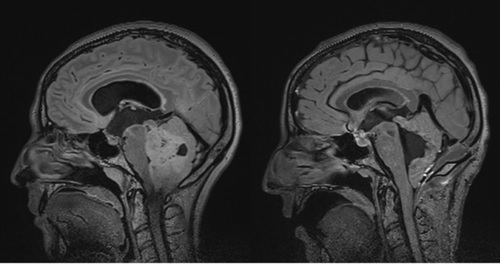
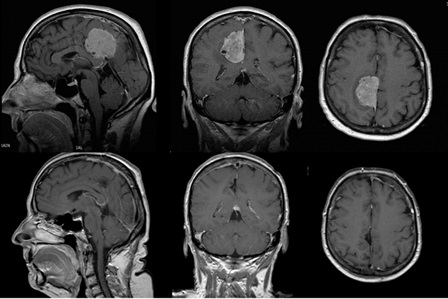
Left frontal lobe astrocytoma
40 years old patient with left frontal lobe astrocytoma. Low grade primary tumours usually are large without causing any particular neurologic symptoms. Diagnosis is usually put following epileptic episodes. The patient remained without any post-operative complications and functional and she returned to work 2 months later.
Postoperatively, in many grade II and in all grade III and IV glioma cases, conventional radiotherapy is administered. In grade III and IV tumours, adjuvant chemotherapy is administered (Temozolamide) during and after radiotherapy. Treatment is sometimes combined with stereotactic radiosurgery, as in the case for smaller foci or relapses.
Meningiomas are the most common benign brain tumours arising from the arachnoid meninge cells. A small percentage of meningiomas develop aggressive behaviour (atypical, anaplastic). Because of their very slow progression, they do not cause acute symptoms and may often be sizeable upon diagnosis. Despite the absence of specific clinical symptoms or signs for brain tumours, the slowly progressing signs and symptoms of increased intracranial pressure (headache, nausea, vomits, somnolence), as well as the manifestation of epilepsy in patients with negative disease history, or motor disturbances, speech and behaviour disorder, should suggest serious suspicion of a developing intracranial lesion.
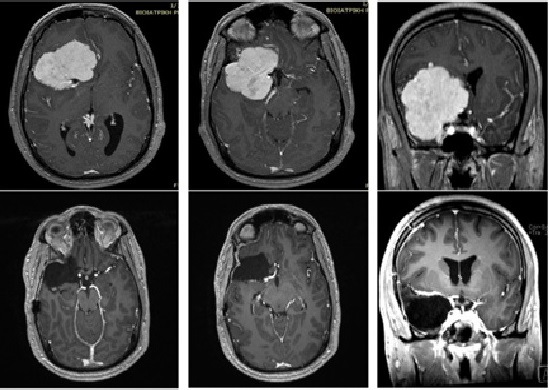
Skull base meningioma
27 years old patient with extensive meningioma, diagnosed following a syncopal episode (probable epileptic seizure). The meningioma has been fully resected through craniotomy.
Thanks to the fact that meningiomas are highly vascularised tumours, they are well imaged with the use of contrast enhanced brain CT scan, paramagnetic substance-enhanced MRI and digital angiography. Their classification is based on their histology type and on the site of the brain where they develop.
Skull base meningioma
38 years old patient with extensive meningioma diagnosed following vision disorders (Right eye visual acuity 3/10, left eye visual acuity 4/10). During the procedure, the entire tunor has been removed and frontal skull base has been reconstructed. Postoperatively, there was normalisation of vision (visual acuity 9/10 bilaterally).
Meningiomas are usually microsurgically excised, leading to cure, if the tumour lies in a surgically accessible site. Many times, highly specific approaches of the skull base are required for their complete resection, accompanied by extensive reconstruction of the skull base. Such operations are usually lengthy and render special post-operative follow-up mandatory.
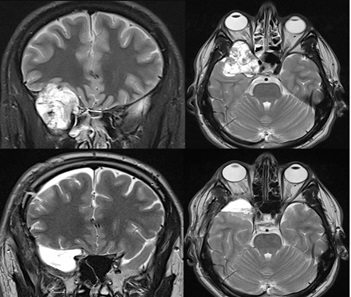
Falx meningioma
58 years old patient with muscle weakness of the lower limb that has been restored post-operatively.
In case of residual tumour or localisation in a difficult to access area, or in cases of histologic atypia / anaplastic meningioma, stereotactic radiosurgery is applied as a treatment of choice.
Atypical meningioma
30 years old patient with extensive meningioma diagnosed following vision disorders (right eye visual acuity 3/10 and diplopia), as well as exophthalmos. During surgery the tumor has been totally removed and the orbita wall was reconstructed.
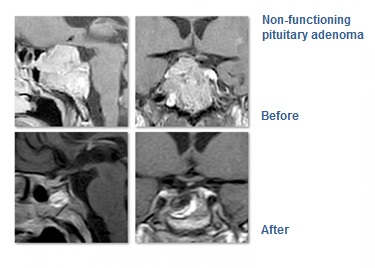
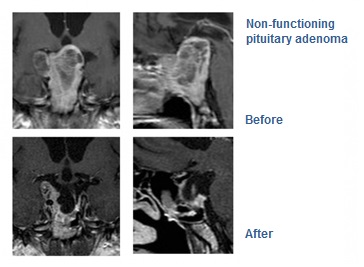
Pituitary adenomas are benign tumours of the pituitary gland and account for approximately 10% of all intracranial neoplasms. They usually remain undiagnosed and such small tumours are found at a percentage of 6 to 24% in the general population.
Symptoms
The diagnosis is based either on visual disorders that are due to compression by the tumour on the optic nerve, or on the symptoms caused by hormones deficiency, due to the affected function of the pituitary gland (pituitary deficiency), or on the clinical symptoms due to excessive hormones secretion. Symptoms’ characteristics depend on the type of hormone that is being hypersecreted.
HORMONE HYPERSECRETION
SYMPTOM
Prolactin (prolactinoma)
Menstrual disorders (dysmenorrhea) or absence of menstruation (amenorrhea), fluid secretion from the breast (galactorrhoea), reduced libido, loss of body hair.
Growth hormone
Acromegaly (increase of upper and lower limbs size), change of facial features, swelling (oedema) of extremities, increase of lower jaw dimensions (prognathism), hyperhidrosis, hypertension, diabetes mellitus)
ACTH (adrenocorticotropic hormone) CUSHING Syndrome
Weight increase, central obesity, moon facies, striae on the abdomen and the breast, reduced muscle mass, skin contusions, acne, hypertrichosis, hypertension, diabetes mellitus, menstrual disorders.
TSH
Basic metabolism increase, excessive sweating, palpitations – tachycardia, increased arterial pressure, increased appetite and concurrent weight loss, high temperature intolerance.
The optic chiasm is the area of the optic tract compressed by specific tumours, causing visual symptoms (bitemporal hemianopsia). In rare cases, they also cause diplopia. Tumours causing visual disorders are usually macroadenomas, larger than 10mm in diameter. Tumours less than 10mm in diameter are called microadenomas. Some tumours secrete more than one hormone, the most frequent combination being growth hormone and prolactin. Prolactinomas are usually diagnosed during pregnancy, when progesterone hormone increases the tumour growth rate. Headaches may be present. Since adenomas are slow to develop, symptoms deterioration may go unnoticed. Only in the case of spontaneous adenoma bleeding (adenoma apoplexy), are the symptoms sudden and always accompanied by headaches, neck pain, nausea, disorders of consciousness and vision disorders.
Diagnosis
The diagnosis is confirmed by checking hormone levels in blood, while neuroimaging methods, e.g. CT scan and mainly MRI play a decisive part. Pre-operative check includes:
- Clinical neurological examination (cranial nerves function test: optic nerve, oculomotor nerve, trochlear nerve and abducens nerve).
- Ophthalmological examination (visual acuity and visual fields tests).
- Imaging – radiology examination (the most significant and absolutely necessary imaging procedure is the pituitary gland MRI with the administration of paramagnetic substance).
- Endocrine system test (complete examination of the pituitary gland function by determining the basic values of the pituitary hormones and by administering stimulation and suppression tests, depending on the clinical picture and the suspected hormonal imbalance, such as suppression of growth hormone through glucose uptake test in acromegaly, cortisol suppression test through the administration of dexamethasone in Cushing’s syndrome).
Management
Pharmaceutical treatment as treatment of choice is applied and is internationally accepted only in the case of prolactinomas. Administration of dopamine agonists (e.g. cabergoline-Dostinex, bromocriptine –Parlodel) enables a significant shrinking of prolactinomas and a long-term control in >95% of cases. There are no similarly effective pharmaceutical treatments for other adenoma types. Particularly in acromegaly (somatostatin analogues – octreotide, lanreotide-, dopamine agonists – bromocriptine, quinagolide, cabergoline – growth hormone receptor antagonist –pegvisomant), in Cushing’s syndrome (ketoconazole-Fungoral, metyrapone, mitotane), as well as in the extremely rare TSH hypersecretion (somatostin analogues, octreotide, lanreotide), the purpose of the pharmaceutical treatment is usually to prepare the patient for surgical operation or to achieve post-operative control of hormonal hypersecretion, in case of failure to treat the endocrine disorder surgically, with or without radiotherapy.
Surgical treatment remains the treatment of choice for all hormone-secreting adenomas (except for prolactinomas), as well as for non-secreting adenomas. Approximately 90% of such tumours are treated through transsphenoidal resection (through the nose). This technique was first reported in 1907 by surgeon Oscar Hirsch in Vienna and has since been significantly improved with the contribution of the father of modern neurosurgery Harvey Cushing (Boston).
TRANSSPHENOIDAL TUMOUR RESECTION
The approach includes a small incision on the nasal mucosa. A path is cleared along the nasal septum, through which access is ensured to the sphenoid sinus. A microscope and/or an endoscope is used to enable visibility of the sella turcica area. Intraoperative fluoroscopy or neuronavigation methods are used in the procedure. In the past few years, endoscopes are widely used for the transnasal approach. In this way, the nasal mucosa is spared and the surgeons are able to have a panoramic view and lighting of the surgical field. The sella turcica floor is released. Following the incision of the basal dura, the surgeon gains access to the adenoma together with the normal pituitary tissue. Selective resection of the mass is performed during which the adenoma is resected whereas the normal tissue remains intact. In certain cases, part of the femoral fascia is used to cover the deficit at the sella turcica, especially in cases of cerebrospinal fluid leak. If the CSF leak is significant, a lumbar drain is temporarily placed. Indications for transsphenoidal resection include: extension of the mass at the sphenoid sinus, infiltration of the sphenoid bone, intrasellar adenomas and symmetrical tumour growth above the sella turcica. This procedure is minimally invasive and allows for immediate patient mobilisation, as soon as anaesthesia wears off. Hospital stay is usually 2 days while rest is recommended for another 10 days at home.
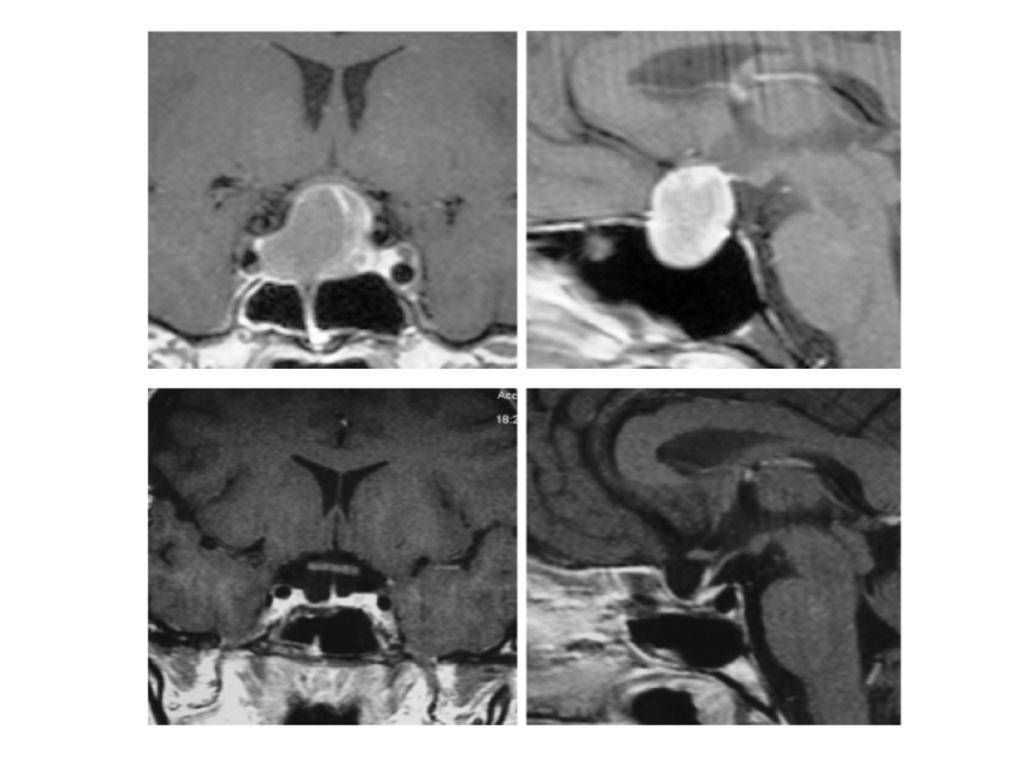
This specific method is not suitable for all pituitary adenomas. Transcranial approaches are sometimes required. The type of procedure is decided on a case by case basis. Tumour characteristics (size, extension beyond the sella turcica) and the patient’s condition (age, general condition and anatomical factors) play a decisive part. Some 10% of cases are not amenable to the transsphenoid technique and the tumour is excised transcranially through a modified ‘mini’ frontotemporal craniotomy. The fissure of Sylvius is accessed, the optic chiasm is prepared together with the arteries of the circle of Willis and the tumour is excised, while healthy pituitary tissue and the vessels remain intact. Indications for transcranial approach include: extension of the tumour subfrontally and/or posteriorly to the optic chiasm, as well as extension towards the middle and/or posterior cranial fossa. At times, other approaches are also applied, such as the transventricular approach for ventricular system tumours in which case a parasagittal craniotomy is performed.
When a cystic tumour puncture or a sella tumour biopsy is required (e.g. for initiation of radiotherapy), the stereotactic method is applied for the approach.
Purpose of surgical procedure
While in the past, the purpose of the procedure was to achieve decompression of the optic nerve and to preserve eye sight, nowadays, thanks to microsurgery, a complete tumour resection is achieved in 70% of patients. Moreover, clinical studies have confirmed that in the hands of experienced surgeons, maintenance of pituitary gland function to pre-operative levels is achieved in more than 70% of patients who undergo the transnasal procedure, while another 20% of patients experience additional improvement. (Nomikos P., Falbusch R, Ladar C., Buchfelder M. Impact of primary surgery on pituitary function in patients with non-functioning pituitary adenomas – a study on 721 patients. Acta Neurochirur (Wien) 146:27-35, 2004).
Invasive tumours, not amenable to complete resection, are treated with postoperative radiotherapy (conventional, fractional radiotherapy or radiosurgery).
Since 2004, Dr. Panagiotis Nomikos and his surgical team at the HYGEIA hospital have treated more than 500 cases of pituitary tumours, combining all modern treatment techniques (endoscopic transnasal surgery, neuronavigation, radiosurgical treatment). This is one of the few pituitary tumours surgical centres, having the full range of equipment and specialisation on this subject, worldwide. The team is also involved in prevention, research and training, actively participating in international pituitary tumours surgery programs.

- © 2007-2025 HYGEIA S.M.S.A.

- Services











